
About the group
We investigate how molecules interact, assemble and crystallize.
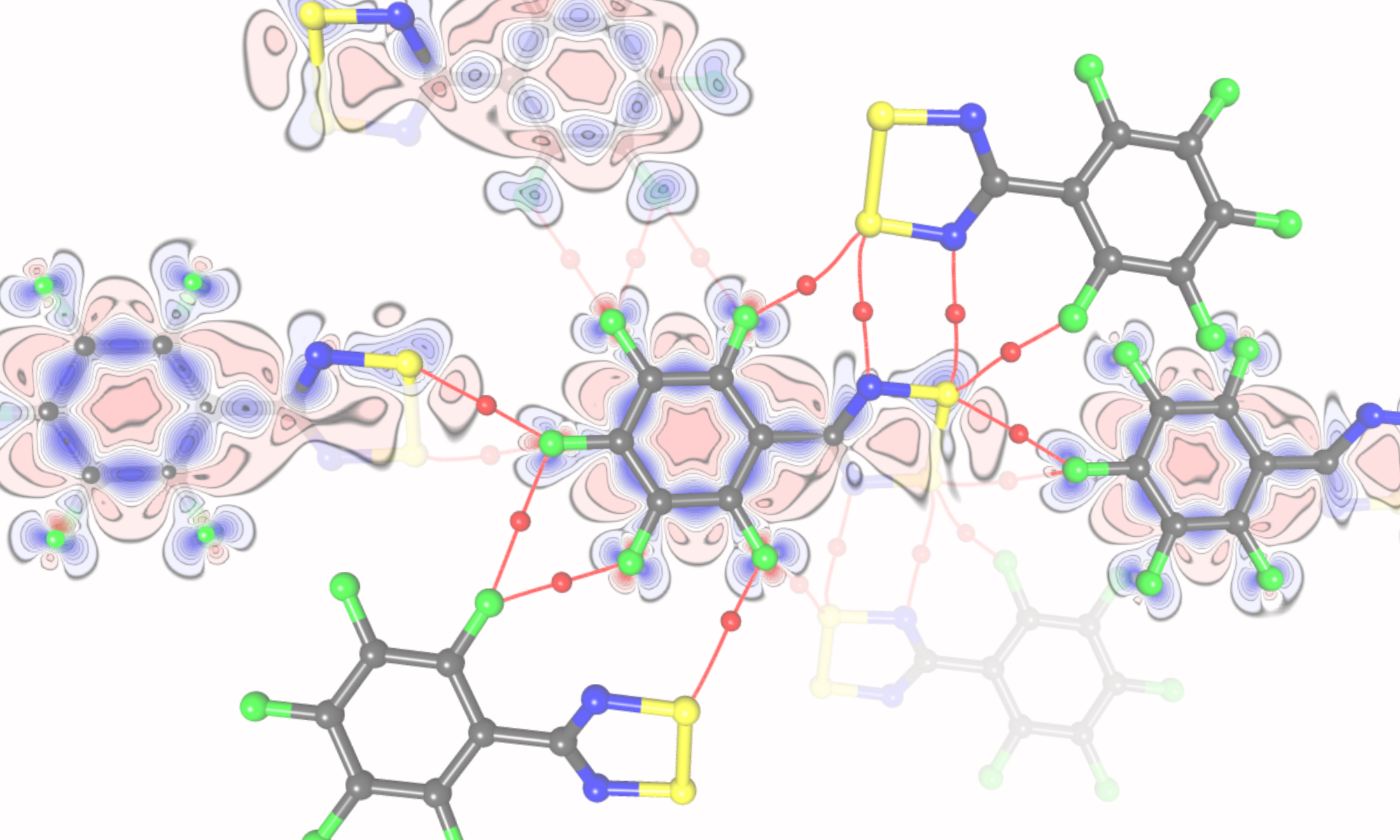
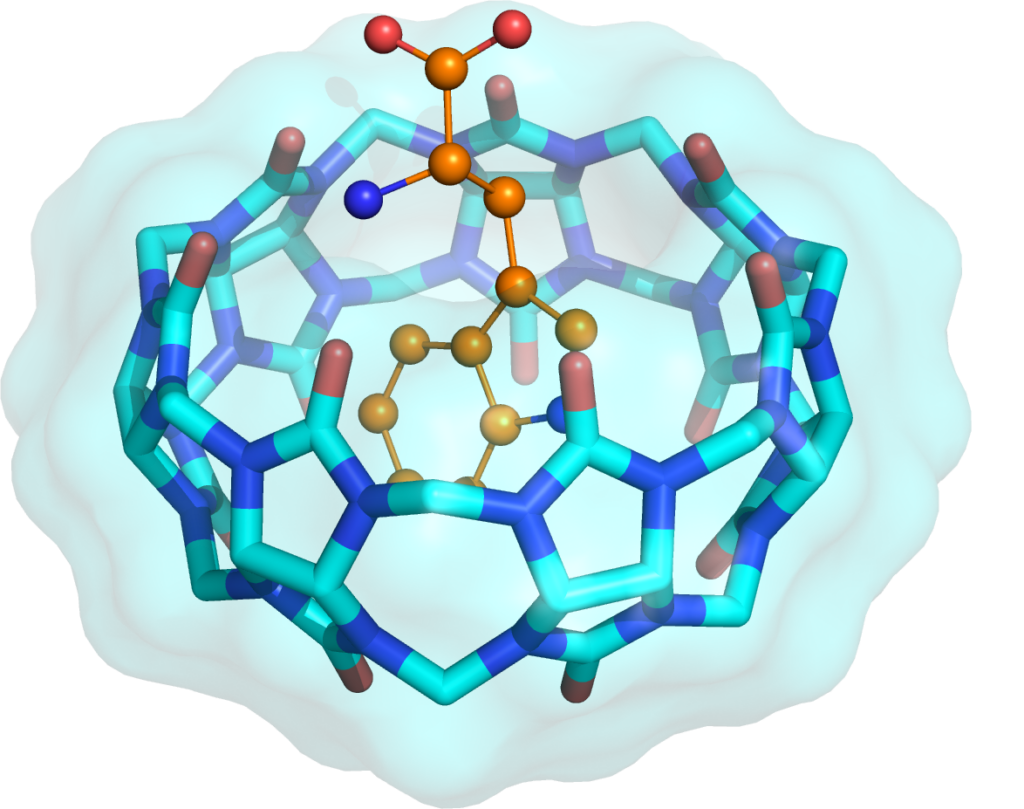
Our group combines high-resolution X-ray diffraction, thermodynamic measurements and computational chemistry to link local non-covalent interactions with global properties such as binding affinity, solubility and crystal morphology. Macrocyclic hosts (calixarenes, cucurbiturils, cavitands) and their complexes with pharmaceutically relevant guests and proteins are central to our work.
Research themes
- Macrocyclic host–guest chemistry in solution and in the solid state
- Crystal growth, polymorphism and morphology of organic and pharmaceutical solids
- Protein–ligand recognition and macrocycles as crystallisation chaperones
- Quantum crystallography and charge-density analysis of biologically relevant systems
- Structure–property relationships and molecular similarity in drug-like molecules
Methods
- Single-crystal X-ray diffraction (laboratory and synchrotron)
- TAAM-based refinement and electron-density reconstruction
- Electrostatic-potential and interaction-energy analyses
- Isothermal titration calorimetry and thermodynamic profiling of binding
- Molecular modelling and molecular dynamics simulations
Projects, publications, opportunities
Projects
- Sonata Bis, 2022-2027, Polish National Science Center,
“True Bioisosteres: Structural and Thermodynamic Classification of Molecular Fragments for Ligand Design” - Sonata, 2017-2020, Polish National Science Center,
“Understanding molecular recognition processes using thermodynamic profiling and structural data “ - Mobility Plus, 2014-2016, Polish Ministry of Science and Higher Education,
“Towards better geometry and charge density of proteins and nucleic acids from synchrotron data” - Etiuda, 2013-2014, Polish National Science Center,
“Structure and Charge Density of Pharmaceutical Substances” - Preludium, 2011-2013, Polish National Science Center,
“Charge Density Studies of Selected Pharmaceutical Substances – Towards Energy of Interactions with Macromolecules “
Selected publications
- Wren, C. P.; Flood, R. J.; Mockler, N. M.; Savko, M.; Malinska, M.; Shi, Q.; Crowley, P. B. Protein Recognition and Assembly by a Phosphocavitand. J. Am. Chem. Soc.2025. https://doi.org/10.1021/jacs.5c08121.
- Wanat, M.; Zaorska, E.; Malinska, M. DMSO Solvates of Tert-Butylcalix[6]Arene and Related Multisolvent Structures. Acta Cryst B 2025, 81 (6). https://doi.org/10.1107/S2052520625008625.
- Pawlędzio, S.; Ziemniak, M.; Wang, X.; Woźniak, K.; Malinska, M. Understanding the Selectivity of Nonsteroidal Anti-Inflammatory Drugs for Cyclooxygenases Using Quantum Crystallography and Electrostatic Interaction Energy. IUCrJ 2025, 12 (2), 208–222. https://doi.org/10.1107/S2052252525000053.
- Zaorska, E.; Malinska, M. Cucurbit[7]Uril-Mediated Histidine Dimerization: Exploring the Structure and Binding Mechanism. Chemistry – A European Journal 2024, n/a (n/a), e202302250. https://doi.org/10.1002/chem.202302250.
- Malinska, M. Insights into Molecular Recognition from the Crystal Structures of P-Tert-Butylcalix[6]Arene Complexed with Different Solvents. IUCrJ 2022, 9 (1), 55–64. https://doi.org/10.1107/S2052252521010678.