
Selected publications:
Zaorska, E.; Stachowicz, M.; Malinska, M. Preferential Crystallization of Tert-Butyl-Calix[6]Arene Chlorobenzene Solvate from a Solvent Mixture. Crystal Growth & Design 2023. https://doi.org/10.1021/acs.cgd.3c00007.
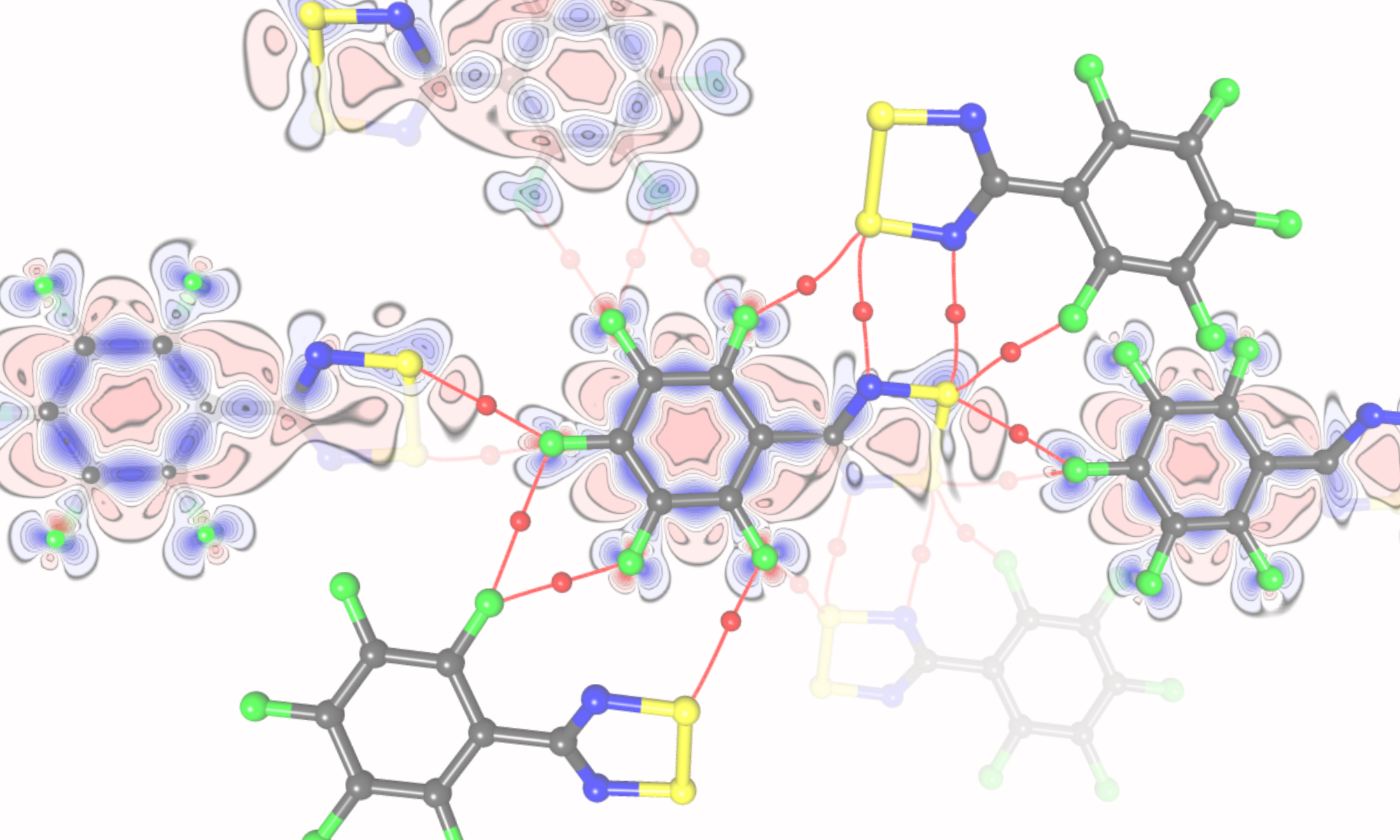
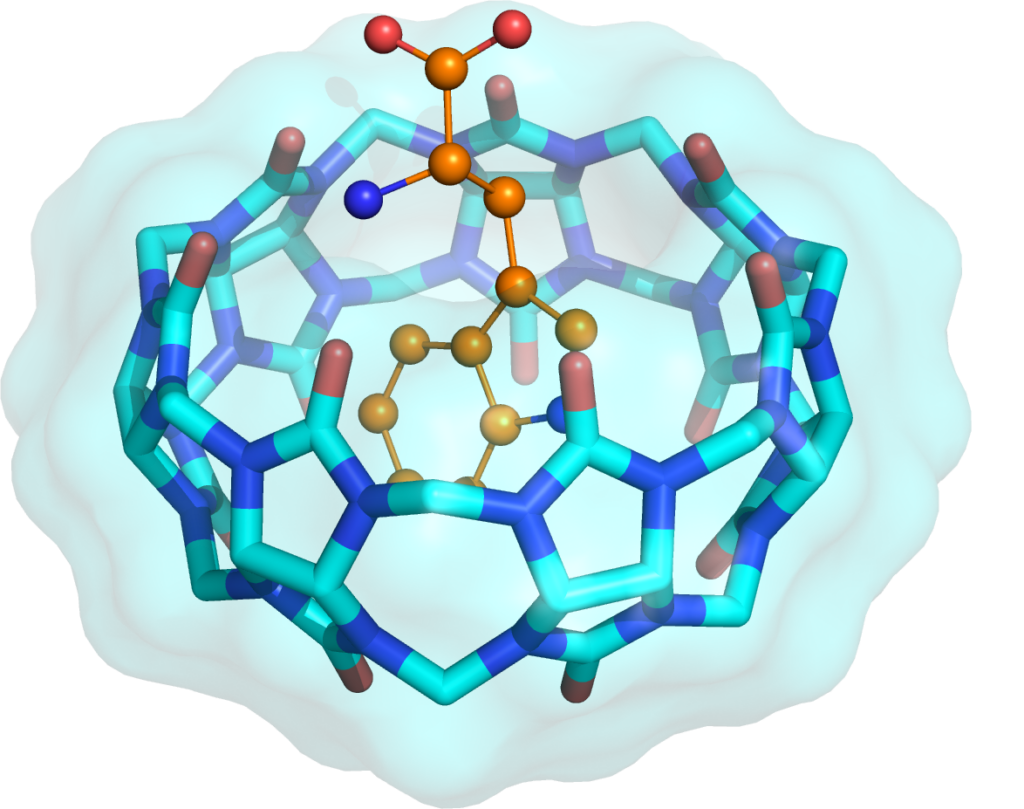
Zaorska, E.; Malinska, M. Cucurbit[7]Uril-Mediated Histidine Dimerization: Exploring the Structure and Binding Mechanism. Chemistry – A European Journal 2023, e202302250. https://doi.org/10.1002/chem.202302250.
Pisarek, J.; Malinska, M. Structure and Morphology of Indole Analogue Crystals. ACS Omega 2020, 5 (28), 17141–17151. https://doi.org/10.1021/acsomega.0c01020.
Malinska, M. Temperature- and Solvent-Induced Crystal-Form Transformations of the Pyridine@p-Tert-Butylcalix[6]Arene Host–Guest System. Cryst. Growth Des. 2021, 21 (2), 1103–1112. https://doi.org/10.1021/acs.cgd.0c01422.
Malinska, M. Insights into Molecular Recognition from the Crystal Structures of p-tert-Butylcalix[6]Arene Complexed with Different Solvents. IUCrJ 2022, 9 (1). https://doi.org/10.1107/S2052252521010678.
Projects:
Sonata Bis, 2022-2027, Polish National Science Center,
“True Bioisosteres: Structural and Thermodynamic Classification of Molecular Fragments for Ligand Design”
Sonata, 2017-2020, Polish National Science Center,
“Understanding molecular recognition processes using thermodynamic profiling and structural data “
Mobility Plus, 2014-2016, Polish Ministry of Science and Higher Education,
“Towards better geometry and charge density of proteins and nucleic acids from synchrotron data”
Etiuda, 2013-2014, Polish National Science Center,
“Structure and Charge Density of Pharmaceutical Substances”
Preludium, 2011-2013, Polish National Science Center,
“Charge Density Studies of Selected Pharmaceutical Substances – Towards Energy of Interactions with Macromolecules “
Current research activities
Medically important macromolecules do not operate as static, isolated moieties. Contrarily, with a remarkable degree of specificity and high affinity, they have numerous interactions with other species, such as proteins, nucleic acids, small molecule ligands, and solvent molecules. Fundamentally, biological processes rely on molecular organization and recognition events. Binding two interacting partners have both enthalpic and entropic components; in other words, the recognition event changes both the structure and dynamics of each counterpart.
Yet, many strategies in medicinal chemistry and crop protection rely on the identification and quantification of molecular similarity. A concept of bioisosterism may be defined as the replacement of a part of a bioactive molecule with a substructure that is similar in size and exhibits similar properties. In the contemporary practice of medicinal chemistry, the development and application of bioisosteres have been adopted as a fundamental tactical approach useful to address a number of aspects associated with the design and development of drug candidates. The established utility of bioisosteres is broad in nature, extending to improving potency, enhancing selectivity, altering physical properties, reducing or redirecting metabolism, eliminating or modifying toxicophores, and acquiring novel intellectual property. The methods of bioisostere identification are limited and fall into two categories, knowledge-based approaches that rely on previously tested fragments that have been substituted for each other without a significant change in the activity under study and computer algorithms that typically score a fragment against the moiety to be replaced using shape or electrostatic measures of similarity. None of these methods takes into account the crucial role of solvation in molecular binding. Therefore, we propose that a group of molecules/molecular fragments may be called bioisosteric if they are all complementary to the same host site in three key elements i.e. steric fit, electrostatic fit, and hydrophobic effect. Only understanding the role of a molecular shape, weak noncovalent interactions, and the role of water in the formation of a complex can provide a more powerful method of prediction of true, diverse bioisosteres.
Biological receptor sites vary greatly in shape, conformational dynamics, and polarity, and require different drug design approaches, therefore simplified model structures are a focus of the project. These synthetic molecules (cucrcubit[7]uril, cucrcubit[7]uril, 4-Sulfocalix[6]arene, 4-Sulfocalix[8]arene) are required to gather reliable information on weak noncovalent and hydrophobic interactions, solvation to classify bioisosteric fragments and define new, more diverse substituents.
Thermodynamic profiling, determining enthalpy and entropy of binding between synthetic ligands and receptors will be analyzed using isotherm titration calorimetry. Through X-ray diffraction, complexes will be crystallized and examined to find interactions responsible for their formation and the location of water molecules. The thermodynamic data will then be analyzed considering the structural data and molecular dynamics results. The molecular dynamic simulations will provide insight into enthalpy and entropy in creating complexes, and water molecule rearrangements during complex formation. Thermodynamic profiling, combined with X-ray structural and computational studies, is the key to elucidate the energetics of the replacement of water by different bioisosteric fragments.
Advances in drug design can lead to significant discoveries, new therapy development, and public health improvement. As medical research improves human health and longevity, the resulting increased productivity may potentially contribute to the national economy in addition to the individual benefits of improved health.